






On May 31, 2021, the NMPA (National Medical Products Administration) announced (No. 76 – 2021) that the newly revised law on the Supervision and Administration of Medical Devices (Order 739) issued by the State Council shall come into force on June 1, 2021. This top-level medical device and IVD law contains a number of important changes, the practical effects of which will be implemented in forthcoming supporting regulations and standards.
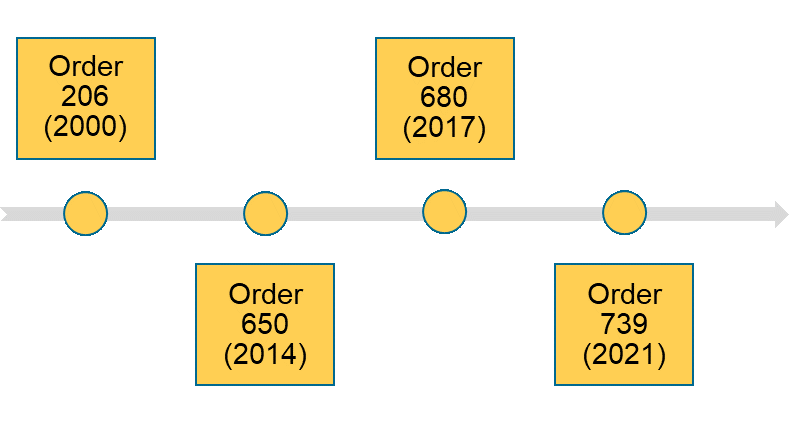
The evolution of China’s Supervision and Administration of Medical Devices
China’s top-level laws are known by the consecutive number that they are passed by the State Council, China’s highest administrative authority. The first top-level law of medical device supervision, Order 206, was passed in the year 2000. More recently, Order 650 was passed in 2014 and revised in 2017 with Order 680.
Order 739 covers a total of 107 articles, an increase of over 25% on the content of the previous Order 680 that was released in 2014 which contained 80 articles. However, the number of chapters in Order 739 and Order 680 remains the same 8 with the same titles as below:
- General Provisions
- Medical Devices Registration and Filing
- Production of Medical Devices
- Distribution and Usage of Medical Devices
- Adverse Events and Recall
- Supervision and Inspection
- Legal Responsibilities
- Supplementary Provisions
Chapters 2, 4, 6 and 7 have received many of the new articles. Existing articles have also been revised and expanded.
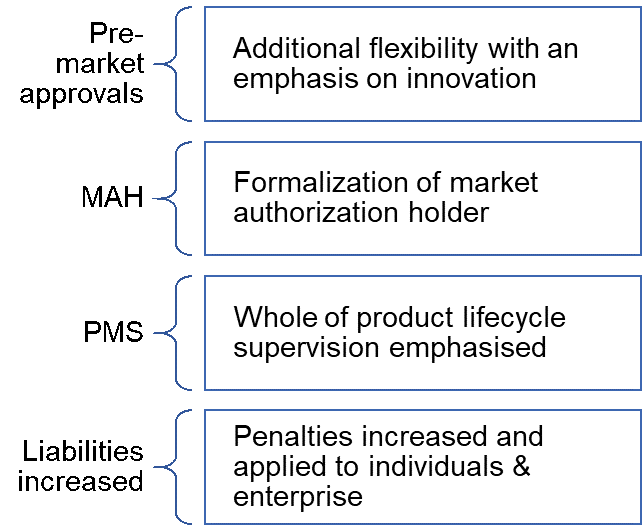
Notable Changes in Order 739
Notable changes in Order 739:
- Increased flexibility in the pre-market approval process with an emphasis on innovation
- “Innovative” devices eligible for faster approval time and dedicated NMPA reviewer throughout the application. Supporting global product registration strategies, medical devices designated as “innovative” no longer require home country approval prior to NMPA application. An innovative device is defined as a ‘product with cutting edge technology, significant clinical application and its own China patent.
- Local China testing requirements can be self-tested by manufacturers instead of being tested in NMPA certified labs. The supporting draft regulation issued on June 1, 2021 clarifies that the overseas manufacturer’s lab will need to be accredited by their relevant government but must follow the on-site verification guidelines of China QMS. However, we strongly caution manufacturers to wait before revising their registration strategies until such process is finally clarified which will take months at least.
- More considered risk-based approach to clinical evaluation so that clinical trials can potentially be avoided even where there is no existing predicate device (or where information relating to the predicate is insufficient to support a clinical evaluation report).
- But where clinical trials are required, a hospital’s capabilities to run them will now be a criterion for their ranking and review. This should greatly incentivise hospitals to increase their trial capacity and expertise. See our post 1,002 Institutions in China Qualified for Clinical Trials for more detail of the facilities now listed on the NMPA database as qualified for hosting clinical trials.
- Lab-developed tests (LDTs) can be used by medical institutions for their own clinical purposes if clinical needs cannot be met by approved IVD reagents. Again, we would like to remind our readers that we need to wait for supporting regulations before changes become tangible.
- Formalization of Market Authorization Holder (MAH) regime
- Until this change design and manufacturing of medical devices and IVDs in China was required to be done by the same entity. The MAH regime has now been formalized for domestic manufacturers but at this stage is not relevant for importing manufacturers.
- Supervision of product lifecycle increased
- There has been a general shift in approach by the NMPA over the last few years from point-in-time registration to whole-of-product lifecycle review. This has now been formalized in Order 739 including with the establishment of a system of professional inspectors. This may increase the frequency of factory inspections going forward, including potentially for overseas factory inspections.
- Mr. Sun Lei, the Director of the CMDE (Center for Medical Device Evaluation), recently published an article addressing the NMPA’s focus on the supervision of product lifecycle. This further helps clarify the regulator’s evolving approach.
- Penalties Increased
- Penalties have been increased by around 50% on entities and key individuals for illegal acts, such as sale of unapproved or non-conforming items. Personal liability is also now introduced on legal representatives, principal responsible persons and other key personnel.
Although the Order 739 is now in force, the practical effect for importing manufacturers is still limited. This is because the subsidiary legislation which clarifies the specific steps and requirements are still in draft.
Therefore, the current regulatory framework is still in a state of flux. As the regulatory measures have not yet been implemented, medical device and IVD registrants should still refer to the 2014 version (as modified in 2017) of the Supervision and Administration of Medical Devices. Manufacturers should keep a close watch on the changes to ensure they remain in compliance and optimize their China registrations.
The following draft regulations concerning the implementation of Order 739 have been released. In our experience, it will be at least a few months or more until these are finalized and in force.
- Technical guidelines for clinical evaluation of medical devices (Draft)
- Technical guidelines for in-vitro diagnostic reagents (Draft)
- Technical guidelines for demonstrating equivalence among medical devices (Draft)
- Technical guidelines for deciding whether to carry out clinical trials of medical devices (Draft)
- Technical guidelines for complaints on clinical evaluation reports concerning medical device registrations (Draft)
- Technical guidelines for determining the methods for conducting clinical trials (Draft)
- Technical guidelines for clinical evaluation reports for medical device registration and filing (Draft)
- Technical guidelines for methods to determine the exemption of clinical trials for in-vitro diagnostic reagents (Draft)
- Clinical trial exemption list for medical devices and in-vitro diagnostic reagents (Draft)
- Dossier submission requirements for the registration of medical devices and the format of document proofs (Draft amendment)
- Dossier submission requirements for the registration of in-vitro diagnostic reagents and the format of document proofs (Draft amendment)
By Hamish King, Jacky Li and Colleen Xu. Contact Cisema if you would like to learn more.


