






Am 31. Mai 2021 gab die NMPA (National Medical Products Administration) bekannt (No. 76 – 2021), dass das vom Staatsrat neu überarbeitete Gesetz zur Überwachung und Verwaltung von Medizinprodukten (Order 739) am 1. Juni 2021 in Kraft treten wird. Dieses übergeordnete Medizinprodukte- und IVD-Gesetz enthält eine Reihe von wichtigen Änderungen, deren praktische Auswirkungen in den kommenden unterstützenden Vorschriften und Normen umgesetzt werden sollen.
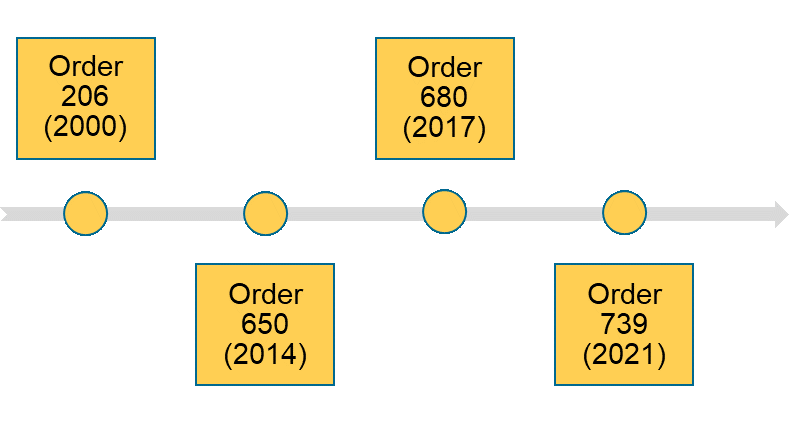
Die Entwicklung von Chinas Gesetzen zur Aufsicht und Verwaltung von Medizinprodukten
Chinas Gesetze der höchsten Ebene werden vom Staatsrat, Chinas höchster Verwaltungsbehörde, verabschiedet und mit fortlaufenden Nummern versehen. Das erste Gesetz dieser Art, Order 206, wurde im Jahr 2000 verabschiedet. In jüngerer Zeit wurde im Jahr 2014 die Order 650 verabschiedet und 2017 mit der Order 680 überarbeitet.
Order 739 umfasst insgesamt 107 Artikel, ein Anstieg von über 25 % gegenüber dem Inhalt der vorherigen Order 680 aus dem Jahr 2014, die 80 Artikel enthielt. Die Anzahl der Kapitel in Order 739 und Order 680 bleibt jedoch die gleiche, nämlich 8 mit den gleichen Titeln wie folgt:
- Allgemeine Bestimmungen
- Registrierung und Einreichung von Medizinprodukten
- Herstellung von Medizinprodukten
- Vertrieb und Anwendung von Medizinprodukten
- Unerwünschte Ereignisse und Rückruf
- Überwachung und Inspektion
- Rechtliche Verantwortlichkeiten
- Ergänzende Bestimmungen
Die Kapitel 2, 4, 6 und 7 haben viele neue Artikel erhalten. Bestehende Artikel wurden ebenfalls überarbeitet und erweitert.
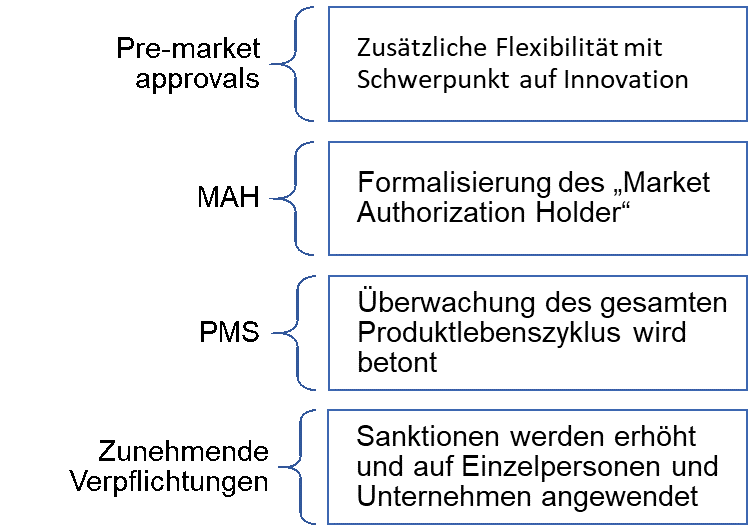
Bemerkenswerte Änderungen in Order 739
Bemerkenswerte Änderungen der Order 739:
- Erhöhte Flexibilität in der Marktzulassung mit Schwerpunkt auf Innovation
- „Innovative“ Produkte haben Anspruch auf eine schnellere Zulassungszeit und einen eigenen NMPA-Prüfer während des gesamten Antragsverfahrens. Zur Unterstützung globaler Produktregistrierungsstrategien benötigen solche Produkte nicht mehr die Genehmigung des Heimatlandes vor dem NMPA-Antrag. Hinweis: Ein innovatives Produkt ist definiert als ein Produkt mit Spitzentechnologie, bedeutender klinischer Anwendung und eigenem China-Patent.
- Lokale China-Testanforderungen können von den Herstellern selbst getestet werden, anstatt in NMPA-zertifizierten Laboren getestet zu werden. Der unterstützende Verordnungsentwurf, der am 1. Juni 2021 veröffentlicht wurde, stellt klar, dass das Labor des ausländischen Herstellers von der jeweiligen Regierung akkreditiert sein muss, aber den Vor-Ort-Verifizierungsrichtlinien des chinesischen QMS folgen muss. Wir raten den Herstellern jedoch dringend, mit der Überarbeitung ihrer Registrierungsstrategien zu warten, bis dieser Prozess endgültig geklärt ist, was mindestens Monate dauern wird.
- Ein überlegterer risikobasierter Ansatz für die klinische Bewertung, so dass klinische Studien möglicherweise auch dann vermieden werden können, wenn es kein existierendes Prädikatsprodukt gibt (oder wenn die Informationen über das Prädikat nicht ausreichen, um den klinischen Bewertungsbericht zu unterstützen).
- Wo jedoch klinische Studien erforderlich sind, wird die Fähigkeit eines Krankenhauses, diese durchzuführen, nun ein Kriterium für ihre Einstufung und Überprüfung sein. Dies sollte ein großer Anreiz für Krankenhäuser sein, ihre Kapazitäten und Expertise für klinische Studien zu erhöhen. Lesen Sie im zugehörigen Blogbeitrag, welche Einrichtungen jetzt in der NMPA-Datenbank für die Durchführung klinischer Studien gelistet sind.
- Laborentwickelte Tests (LDTs) können von medizinischen Einrichtungen für ihre eigenen klinischen Zwecke verwendet werden, wenn der klinische Bedarf nicht durch zugelassene IVD-Reagenzien gedeckt werden kann. Wir möchten unsere Leser noch einmal darauf hinweisen, dass wir auf unterstützende Regelungen warten müssen, bevor Änderungen greifbar werden.
- Formalisierung des Market Authorization Holder (MAH) Systems
- Bisher war es in China erforderlich, dass Design und Herstellung von Medizinprodukten und IVDs von der gleichen Einrichtung durchgeführt wurden. Das MAH-Regime wurde nun für inländische Hersteller formalisiert – dies gilt nicht für importierende Hersteller.
- Überwachung des Produktlebenszyklus verstärkt
- In den letzten Jahren hat sich der Schwerpunkt von der Marktzulassung vor dem Inverkehrbringen auf den gesamten Produktlebenszyklus verlagert. Dies wurde nun in der Order 739 formalisiert, u. a. durch die Einrichtung eines Systems professioneller Inspektoren. Dies könnte die Häufigkeit von Werksinspektionen (auch im Ausland) in Zukunft erhöhen.
- Herr Sun Lei, der Direktor des CMDE (Center for Medical Device Evaluation), hat kürzlich einen Artikel veröffentlicht, der den Fokus der NMPA auf die Überwachung des Produktlebenszyklus anspricht. Dies hilft, den sich entwickelnden Ansatz der Regulierungsbehörde zu verdeutlichen.
- Bußgelder wurden erhöht
- Die Strafen für illegale Handlungen, wie z.B. den Verkauf von nicht zugelassenen oder nicht konformen Produkten, wurden für Unternehmen und Schlüsselpersonen um ca. 50% erhöht. Die persönliche Haftung wird nun auch für gesetzliche Vertreter, Hauptverantwortliche und andere Schlüsselpersonen eingeführt.
Obwohl die Order 739 nun in Kraft ist, ist die praktische Auswirkung für importierende Hersteller noch begrenzt. Dies liegt daran, dass die untergeordneten Gesetze, die die spezifischen Schritte und Anforderungen klarstellen, noch entworfen werden.
Daher befindet sich der derzeitige Rechtsrahmen noch im Fluss. Da die regulatorischen Maßnahmen noch nicht umgesetzt werden, sollten sich Medizinprodukte- und IVD-Antragsteller weiterhin auf die 2014er Version (mit Änderungen von 2017) der Supervision and Administration of Medical Devices beziehen. Hersteller sollten die Änderungen genau im Auge behalten, um sicherzustellen, dass sie konform bleiben und ihre Registrierungen in China optimieren.
Folgende Regelungsentwürfe zur Umsetzung der Order 739 wurden bisher veröffentlicht. Unserer Erfahrung nach wird es noch einige Monate oder mehr dauern, bis diese fertiggestellt und in Kraft sind.
- Technische Leitlinien für die klinische Bewertung von Medizinprodukten (Entwurf)
- Technische Leitlinien für In-Vitro-Diagnostik Reagenzien (Entwurf)
- Technische Leitlinien für den Nachweis der Äquivalenz von Medizinprodukten (Entwurf)
- Technische Leitlinien für die Entscheidung über die Durchführung von klinischen Studien von Medizinprodukten (Entwurf)
- Technische Leitlinien für Beanstandungen von klinischen Bewertungsberichten bei Medizinproduktezulassungen (Entwurf)
- Technische Leitlinien für die Festlegung der Methoden zur Durchführung von klinischen Studien (Entwurf)
- Technische Leitlinien für Berichte zur klinischen Bewertung bei der Registrierung und Notifizierung von Medizinprodukten (Entwurf)
- Technische Leitlinien für Methoden zur Festlegung der Freistellung von klinischen Studien für In-Vitro-Diagnostik Reagenzien (Entwurf)
- Ausnahmeliste für klinische Studien für Medizinprodukte und In-Vitro-Diagnostik Reagenzien (Entwurf)
- Anforderungen an die Einreichung von Registrierungsdossiers von Medizinprodukten und das Format von Dokumentennachweisen (Änderungsentwurf)
- Anforderungen an die Einreichung von Registrierungsdossiers von In-Vitro-Diagnostik Reagenzien und das Format der Dokumentennachweise (Änderungsentwurf)
Von Hamish King, Jacky Li and Colleen Xu. Kontaktieren Sie Cisema, wenn Sie mehr erfahren möchten.

")
")